DNA origami
DNA origami folding
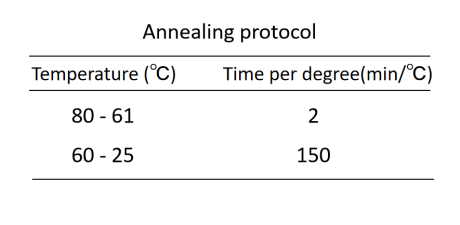
DNA origami structures were designed utilizing the cadnano software and folded by mixing a final concentration of 10 nM p7249 scaffold, 100 nM each of the staple strands, 1x TE buffer (containing 10 mM Tris, 1 mM EDTA, pH 8) and 4 mM MgCl2. A total volume of 25 μL of this mixture was subjected to annealing in a PCR instrument to facilitate the proper folding of the DNA origami structures. The annealing protocol is as follows:
DNA origami purification
To remove excess staple strands, 100 µL of the annealed DNA origami solution was diluted with 1x TE buffer and 20 mM MgCl2 to a final volume of 500 µL, and then mixed with PEG purification solution (15% w/v PEG-8000, 500 mM NaCl, and 1x TE) in a 1:1 volume ratio, waiting 10 min for the samples to equilibriate and then centrifuging at 20,000 RCF and 4℃for 30 min. After centrifugation, the supernatant was carefully removed, and the pellet was resuspended in 20 µL resuspension buffer (1x TE buffer and 4 mM MgCl2) by shaking it overnight at 37℃with a shaking speed of 200 rpm on an shaker. After re-suspension, the DNA concentration was measured with a Nanodrop and adjusted to 12 nM concentration of DNA origami.
AuNR
DNA modification
Thiol DNA was prepared into a 100 µM concentration aqueous solution. Take 20 µL of thiol DNA solution into an EP tube, add 4 µL of 1 M TCEP solution, 2.6 µL of distilled water, and let it stand at room temperature for 1 hour. Afterwards, use a pipette to transfer the mixture into 4.46 mL of gold nanorod solution, mix evenly, and incubate at room temperature for 24 hours. After the reaction is complete, centrifuge the solution at 3000 rpm for about 15 minutes. Once the centrifugation is complete, use a pipette to remove the supernatant and resuspend it in 150 µL distilled water to obtain surface thiol DNA modified gold nanorods.
Doara
DNA origami & AuNP assembly
AuNR and DNA origami were mixed at a molar ratio of 3:2, then the mixture was annealed from 37 °C to room temperature in water bath overnight.
Characterization
Agarose gel electropherosis
1% agarose gel was prepared by mixing 50 mL 1x TAE buffer, 0.5 g agarose, and 200 μL 1 M MgCl2. The gel was run in 1x TAE buffer containing 4 mM Mg2+ at 60 V and 4°C for two hours.
TEM sample preparation
Unstained sample: The preparation of unstained samples involved treating copper grids with a plasma cleaner for 25 seconds, followed by dropping 5 μL sample to the grids and left to dry. The samples were imaged with a TEM operating at 100 kV.
Negative stain sample: The preparation of negatively stained samples involved treating copper grids with a plasma cleaner for 25 seconds. Then drop 5 μL sample to the grids and left to dry. Subsequently, 5 μL of 2% uranyl acetate was dropped to the grids for negative staining for 2 minutes. The excess stain was wicked away with filter paper, and the grids were left to dry. The samples were imaged with a TEM operating at 100 kV.
Zeta potential
Transfer 20 µL of the test solution into a 1.5 mL EP tube, and dilute with distilled water to 1000 µL. After mixing evenly, use a pipette to transfer 700 µL into a Malvern Zetasizer Nano Series and then place it in a Malvern Nanoparticle Size and Zeta Potential Analyzer. Scan time 30 seconds, scan 3 times, start testing.
Dynamic light scattering
Transfer 20 µL of the test solution into a 1.5 mL EP tube, and dilute with distilled water to 1000 µL. After mixing evenly, all samples were transferred into Malvern Glass Cuvettes and loaded into the Nanoparticle Size and Zeta Potential Analyzer. Scan time 30 seconds, scan 3 times, start testing.
UV-Vis spectroscopy
Use a pipette to take 50 µL of the test solution, place it in a 50 µL High Precision Cell, and then place it in the UV-Vis-NIR Spectrometer.Set the parameters: scan wavelength from 1100 nm to 400 nm, scan interval of 2 nm, use distilled water as a reference, and start scanning.